

Fibrosi quística
La fibrosi quística és una malaltia hereditària, degenerativa i potencialment greu. Aproximadament, un 15% dels nadons amb fibrosi quística presenten símptomes en el moment del naixement en forma d'obstrucció intestinal.
Què es la fibrosi quística?
La fibrosi quística és una malaltia hereditària, degenerativa i potencialment greu, que afecta diversos òrgans i sistemes del cos, especialment els pulmons i el sistema digestiu.
Aquesta malaltia és autosòmica recessiva, per la qual cosa, s'ocasiona si la persona té una mutació a les dues còpies del gen CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), situat al cromosoma 7.
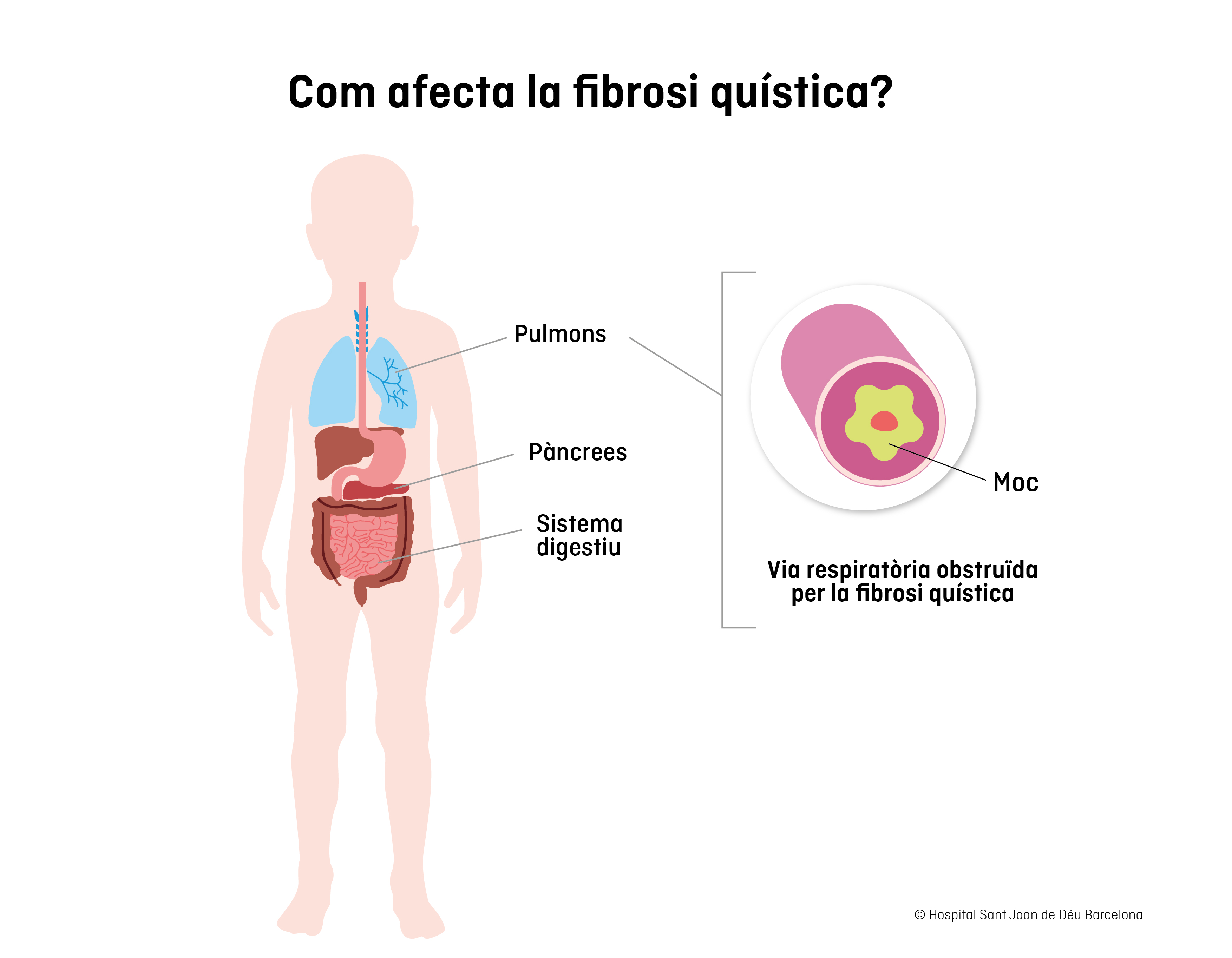
Aquest gen produeix una proteïna que té el mateix nom (pCFTR ) i actua com a canal de clorur, regulant la quantitat d'aigua i sal que contenen els líquids interns de l'organisme. Quan el gen està afectat, les secrecions són més viscoses, el moc més espès i enganxós i, de manera progressiva, es van obstruint o tapant els conductes respiratoris, pancreàtics i intestinals, ocasionant l'afectació dels òrgans.
La prevalença de la fibrosi quística és variable segons la zona geogràfica. En general, s'estima que al nostre entorn afecta aproximadament un de cada 6.500 nadons vius i que, aproximadament, una de cada 35 persones és portadora d'una sola mutació (i, per tant, no presenta la malaltia).
Quins són els seus símptomes?
El tipus i la intensitat dels símptomes de la fibrosi quística són molt variables i depenen de diversos factors: les mutacions genètiques del pacient, l'edat que té quan se'l diagnostica, l'accés a tractament modulador de CFTR, l’emplenament dels diferents tractaments i els factors socioambientals, entre d'altres.
Aproximadament, un 15% dels nadons amb fibrosi quística presenten símptomes en el moment del naixement en forma d'obstrucció intestinal. Aquesta afectació impedeix l'eliminació de les primeres femtes del nadó, i pot provocar vòmits, inflor abdominal, dolor i complicacions que poden ser greus. El 85% dels casos, ja des del naixement, presenta problemes per augmentar de pes adequadament, ja que l'absorció dels nutrients ja està afectada.
Altres símptomes habituals a la primera infància són tosos persistent, infeccions respiratòries i/o bronquitis recurrents, diarrea crònica, dificultat per respirar i sudoració excessivament salada, tenint més risc de deshidratació.
En els nens grans, adolescents i adults, els símptomes poden incloure:
-
Tos crònica
-
Infeccions respiratòries recurrents
-
Diferents complicacions respiratòries
-
Fatiga
-
Mala absorció de nutrients amb diarrea crònica i/o retard en pes i de creixement
-
Diabetis
-
Pòlips nasals i secreció de la mucosa nasal de forma habitual
-
Problemes de fetge
-
Problemes de fertilitat als barons
Com es diagnostica?
El diagnòstic de la fibrosi quística es realitza per cribratge neonatal a partir d'una mostra de sang extreta per punció al taló entre les 48 i les 72 hores de vida.
Un cribratge neonatal positiu de fibrosi quística no significa que el pacient tingui la malaltia, sinó que té més probabilitat de patir la malaltia que la població general i, per tant, cal fer el diagnòstic que ho confirmi en una Unitat de Fibrosi Quística de referència.
Aquest diagnòstic confirmatori es basa en el test de la suor (anàlisi de la suor del nadó per quantificar la quantitat de clorur que té) i l’estudi genètic (per identificar cada mutació en les dues còpies del gCFTR).
Molts dels nadons a què es realitza un cribratge neonatal no pateixen fibrosi quística i l'estudi que es realitza després a la Unitat de Fibrosi Quística de referència descarta l'existència de malaltia. Es diu en aquests casos, que el programa de cribratge neonatal de fibrosi quística és molt sensible (hi ha gran probabilitat que s'identifiquin els nadons amb la malaltia) però poc específic (es pot identificar un nadó com a malalt quan, en realitat, no ho està).
Quin és el seu tractament?
El tractament clàssic de la fibrosi quística es basa a pal·liar (o millorar) els símptomes i complicacions de la malaltia, sent imprescindible per millorar el pronòstic i la qualitat de vida dels pacients. Els tractaments han de ser individualitzats, però habitualment es recomana a la majoria dels pacients:
-
Teràpia d'aerosol per ajudar a eliminar el moc dels pulmons
-
Antibiòtics per tractar les infeccions respiratòries
-
Enzims pancreàtics per ajudar a la digestió
-
Suplements nutricionals per millorar la nutrició
-
Exercici físic i fisioteràpia per millorar la capacitat pulmonar
En els darrers anys han aparegut noves opcions terapèutiques d'alta efectivitat per al tractament de la malaltia: els tractaments moduladors de CFTR. Aquests fàrmacs corregeixen la funció i/o quantitat de pCFTR activa i, per tant, actuen com a tractament de la malaltia.
Els tractaments moduladors de CFTR estan indicats per a unes mutacions concretes i se n'ha autoritzat l'ús a partir de determinades edats, segons el cas. Aquestes teràpies han ocasionat una gran millora en la qualitat de vida, el pronòstic i la supervivència dels pacients.
Quines altres coses cal tenir en compte?
És important que els nens amb fibrosi quística rebin un diagnòstic primerenc i un tractament adequat i individualitzat per poder viure amb la màxima qualitat de vida possible, amb menys símptomes i per prevenir complicacions greus.
Atesa la complexitat de la malaltia, és imprescindible que els pacients siguin atesos a una Unitat de Fibrosi Quística específica, amb un programa estandarditzat d'atenció i tractament multidisciplinari, amb especialistes coordinats pels professionals de referència.


