

Fibrosis quística
La fibrosis quística es una enfermedad hereditaria, degenerativa y potencialmente grave. Aproximadamente un 15% de los recién nacidos con fibrosis quística presentan síntomas en el momento del nacimiento en forma de obstrucción intestinal.
¿Qué es la fibrosis quística?
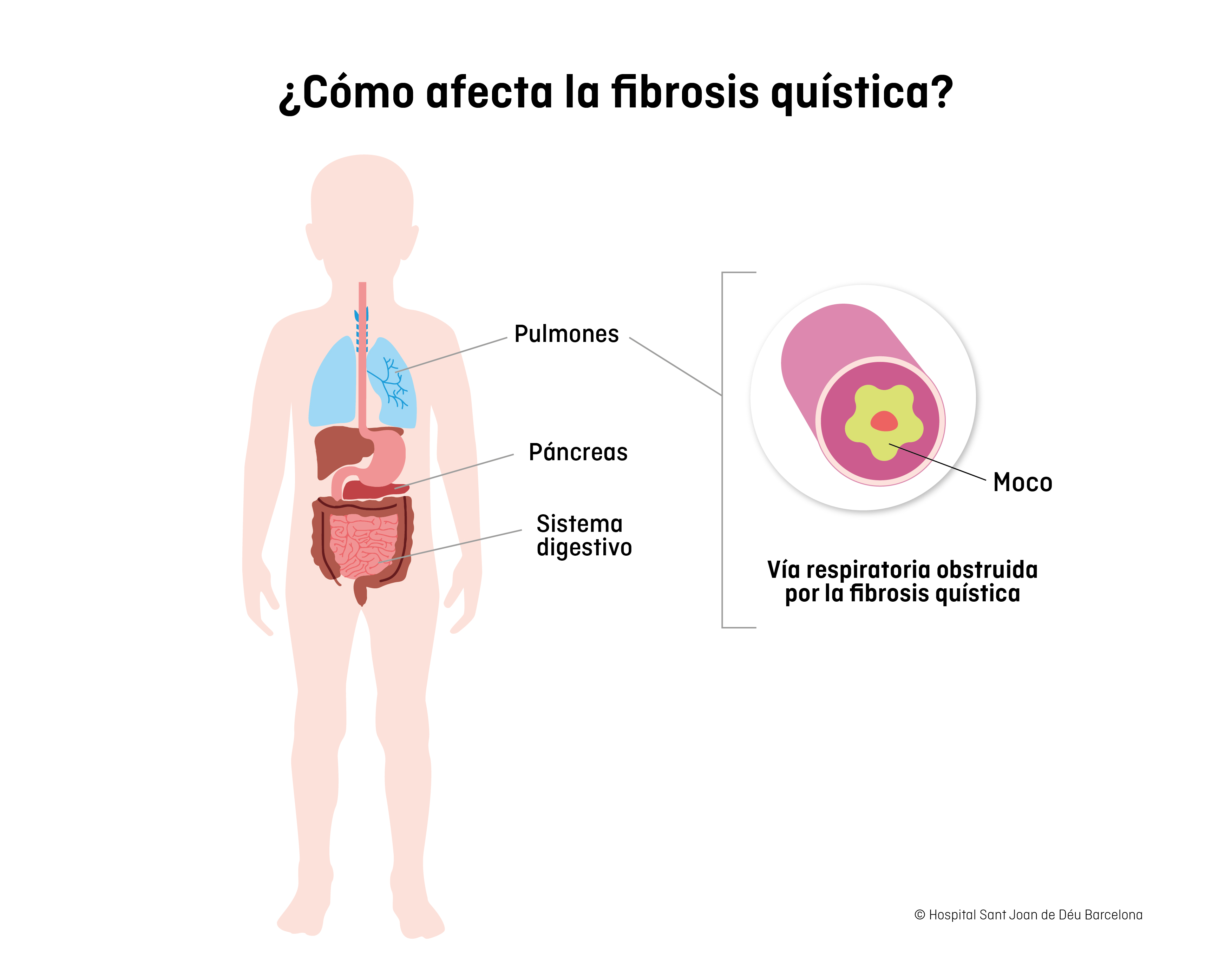
La fibrosis quística es una enfermedad hereditaria, degenerativa y potencialmente grave, que afecta a varios órganos y sistemas del cuerpo, especialmente a los pulmones y al sistema digestivo.
Esta enfermedad es autosómica recesiva, por lo que se ocasiona si la persona tiene una mutación en las dos copias del gen CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), situado en el cromosoma 7.
Este gen produce una proteína que tiene el mismo nombre (pCFTR) y actúa como canal de cloruro, regulando la cantidad de agua y sal que contienen los líquidos internos del organismo. Cuando el gen está afectado, las secreciones son más viscosas, el moco más espeso y pegajoso y, de forma progresiva, se van obstruyendo o tapando los conductos respiratorios, pancreáticos e intestinales, ocasionando la afectación de los órganos.
La prevalencia de la fibrosis quística es variable según la zona geográfica. En general, se estima que en nuestro entorno afecta aproximadamente a uno de cada 6.500 recién nacidos vivos y que, aproximadamente, una de cada 35 personas es portadora de una sola mutación (y, por lo tanto, no presenta la enfermedad).
¿Cuáles son sus síntomas?
El tipo y la intensidad de los síntomas de la fibrosis quística es muy variable y dependen de varios factores: las mutaciones genéticas del paciente, la edad que tiene cuando se le diagnostica, el acceso a tratamiento modulador de CFTR, la cumplimentación de los distintos tratamientos y factores socioambientales, entre otros.
Aproximadamente un 15% de los recién nacidos con fibrosis quística presentan síntomas en el momento del nacimiento en forma de obstrucción intestinal. Esto impide la eliminación de las primeras heces del bebé, y pueden aparecer vómitos, hinchazón abdominal, dolor y complicaciones que pueden ser graves. El 85% de los casos, ya desde el nacimiento, presenta problemas para aumentar de peso adecuadamente, pues la absorción de los nutrientes ya está afectada.
Otros síntomas habituales en la primera infancia son tos persistente, infecciones respiratorias y/o bronquitis recurrentes, diarrea crónica, dificultad para respirar y sudoración excesivamente salada, teniendo mayor riesgo de deshidratación.
En los niños mayores, adolescentes y adultos, los síntomas pueden incluir:
-
Tos crónica
-
Infecciones respiratorias recurrentes
-
Diferentes complicaciones respiratorias
-
Fatiga
-
Mala absorción de nutrientes con diarrea crónica y/o retraso en peso y de crecimiento
-
Diabetes
-
Pólipos nasales y secreción de la mucosa nasal de forma habitual
-
Problemas de hígado
-
Problemas de fertilidad en los varones
¿Cómo se diagnostica?
El diagnóstico de la fibrosis quística se realiza por cribaje neonatal a partir de una muestra de sangre extraída por punción en el talón entre las 48 y las 72 horas de vida.
Un cribaje neonatal positivo de fibrosis quística no significa que el paciente tenga la enfermedad, sino que tiene mayor probabilidad de sufrir la enfermedad que la población general y, por lo tanto, debe realizarse el diagnóstico que lo confirme en una Unidad de Fibrosis Quística de referencia.
Este diagnóstico confirmatorio se basa en el test del sudor (análisis del sudor del bebé para cuantificar la cantidad de cloruro que tiene) y el estudio genético (para identificar cada mutación en las dos copias del gCFTR).
Muchos de los bebés a los que se realiza un cribaje neonatal no sufren fibrosis quística y el estudio que se realiza después en la Unidad de Fibrosis Quística de referencia descarta la existencia de enfermedad. Se dice, en estos casos, que el programa de cribaje neonatal de fibrosis quística es muy sensible (hay gran probabilidad de que se identifique a los bebés con la enfermedad) pero poco específico (se puede identificar a un bebé como enfermo cuando, en realidad, no lo está).
¿Cuál es su tratamiento?
El tratamiento clásico de la fibrosis quística se basa en paliar (o mejorar) los síntomas y complicaciones de la enfermedad, siendo imprescindible para mejorar el pronóstico y la calidad de vida de los pacientes. Los tratamientos deben ser individualizados, pero habitualmente se recomienda en la mayoría de los pacientes:
-
Terapia de aerosol para ayudar a eliminar el moco de los pulmones
-
Antibióticos para tratar las infecciones respiratorias
-
Enzimas pancreáticas para ayudar a la digestión
-
Suplementos nutricionales para mejorar la nutrición
-
Ejercicio físico y fisioterapia para mejorar la capacidad pulmonar
En los últimos años, han aparecido nuevas opciones terapéuticas de alta efectividad para el tratamiento de la enfermedad: los tratamientos moduladores de CFTR. Estos fármacos corrigen la función y/o cantidad de pCFTR activa y, por lo tanto, actúan como tratamiento de la enfermedad.
Los tratamientos moduladores de CFTR están indicados para unas mutaciones concretas y se ha autorizado su uso a partir de determinadas edades, según el caso. Estas terapias han ocasionado una gran mejoría en la calidad de vida, el pronóstico y la supervivencia de los pacientes.
¿Qué otras cosas hay que tener en cuenta?
Es importante que los niños con fibrosis quística reciban un diagnóstico temprano y un tratamiento adecuado e individualizado para poder vivir con la mayor calidad de vida posible, con menos síntomas y para prevenir complicaciones graves.
Dada la complejidad de la enfermedad, es imprescindible que los pacientes sean atendidos en una Unidad de Fibrosis Quística específica, con un programa estandarizado de atención y tratamiento multidisciplinario, con especialistas coordinados por los profesionales de referencia.


