

Deficiència d'Ornitina Transcarbamilasa (OTC)
La deficiència d’Ornitina Transcarbamilasa (OTC) afecta el cicle de la urea i produeix hiperamonèmia (nivells alts d’amoníac a la sang). La causa és una mutació al gen OTC que es troba al cromosoma X. Això vol dir que, tant l’herència com la clínica tindran unes característiques diferents entre homes i dones.
Què és la Deficiència d'Ornitina Transcarbamilasa?
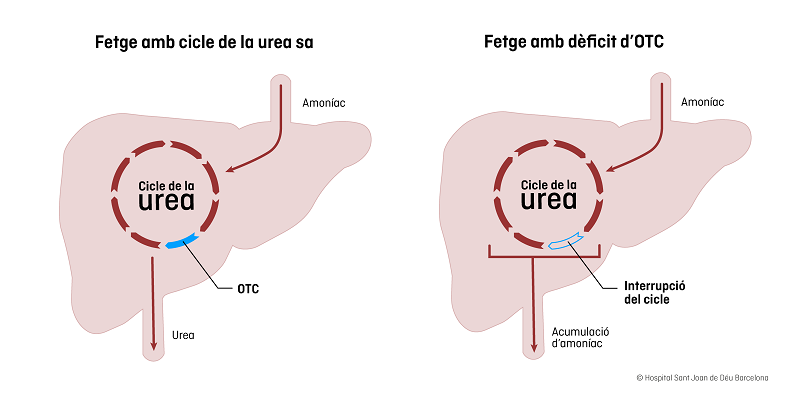
L’Ornitina Transcarbamilasa (OTC) és un enzim que participa al cicle de la urea i s’expressa (o sintetitza proteïnes) majoritàriament al fetge i, en molt petita proporció, al duodè.
El cicle de la urea és el procés que converteix l'amoniac tòxic en urea i s'elimina per l'orina. La deficiència d’OTC afecta el cicle de la urea i produeix hiperamonèmia (nivells alts d’amoníac a la sang). La causa és una mutació al gen OTC que es troba al cromosoma X. Això vol dir que, tant l’herència com la clínica tindran unes característiques diferents entre homes i dones.
Com s'hereda el gen OTC?
Si la mare és portadora d’una mutació al gen OTC, pot patir, o no, els efectes de la deficiència en aquest enzim. Si té un fill home, la dona transmetrà la mutació al seu fill (passant-li el cromosoma X afectat) i, com els homes només tenen un cromosoma X i aquest té un gen OTC mutat, el nen tindrà la malaltia.
En el cas de les filles d’aquesta dona, com les dones tenen dos cromosomes X, la filla serà portadora i podrà transmetre el gen afectat al seus propis fills. És a dir, la meitat de les filles d’una dona portadora seran també portadores i la meitat dels fills tindran la malaltia.

Respecte el pare, si pot tenir descendència, transmetrà el gen mutat a les seves filles (a totes) amb el seu cromosoma X. En canvi, els seus fills seran tots sans.

També poden haver mutacions espontànies, que no es transmeten per via materna.
Quins són els seus símptomes?
Els nens tenen un cromosoma X (de la mare) i un cromosoma Y (del pare). Si el gen OTC del cromosoma X està afectat, la seva activitat enzimàtica també estarà afectada. En aquest cas, els símptomes de la deficiència d’OTC dependran de la magnitud d’aquesta deficiència. Els nens poden tenir una forma neonatal greu de la malaltia (tot i que poden haver casos més lleus, de presentació posterior, a qualsevol etapa de la vida).
En el cas de les nenes, com que tenen dos cromosomes X, si un està afectat, quedarà aproximadament la meitat de l’activitat enzimàtica del gen. Tanmateix, durant el desenvolupament dels cromosomes X, un d’ells (el normal o l’afectat) s’inactiva de forma aleatòria. Per tant, si és el gen afectat el que continua actiu, el defecte d’OTC es veurà sobre tot al fetge (què és la part del cos a on més s’expressa el gen) i la nena tindrà més o menys símptomes depenent de la quantitat de cèl·lules hepàtiques que tingui el gen afectat actiu.
La simptomatologia de la deficiència d’OTC serà, doncs, variable. Es pot manifestar des del naixement, a la infància, l’adolescència o a l’edat adulta. La presentació tardana pot aparèixer en forma de dificultats cognitives, vòmits recurrents, hepatopatía, aversió a les proteïnes i, fins i tot, crisis abruptes (brusques, repentines) de trastorns de conducta. A les dones és més freqüent que hi hagi formes cròniques lleus, amb símptomes com la dificultat en els aprenentatges i símptomes digestius (anorèxia, intolerància a les proteïnes). També poden haver casos gairebé asimptomàtics.
Com es diagnostica?
El diagnòstic de la deficiència d’OTC es realitza d’acord amb la sospita clínica. En períodes de descompensació metabòlica, s’han de mesurar en una analítica l’amoni, els aminoàcids i l’àcid oròtic.
La confirmació del diagnòstic de la deficiència d’OTC es realitza amb un estudi genètic que permetrà realitzar consell genètic i diagnòstic prenatal.
Quin és el tractament?
El tractament de deficiència d’OTC és similar al que es fa a la resta de defectes del cicle de la urea. La base és el tractament dietètic amb restricció proteica de manera persistent per evitar l’acumulació d’amoni. A més a més, hi ha fàrmacs que permeten depurar aquest amoni i reduir els seus nivells en sang. D'altra banda, en crisis de descompensació amb nivells d’amoni molt elevats, s’ha de fer una depuració urgent per mètodes d'hemodiàlisi o hemofiltració.
També es pot valorar el trasplantament hepàtic en pacients amb deficiència d’OTC greu d'inici neonatal (que, en general, es realitza entre els 3 i els 6 mesos d'edat) o en els pacients amb difícil control metabòlic.
Quines altres coses cal tenir en compte?
És important realitzar el diagnòstic de les dones portadores perquè, tot i que poden estar sense símptomes, també poden aparèixer de forma sobtada, posant en perill la seva vida.
D'altra banda, és important fer estudis familiars ja que poden haver altres membres de la família afectats (homes i dones) amb possibilitat de debutar de manera tardana amb quadres molt greus.
La detecció precoç pot evitar aquestes presentacions. El cribatge neonatal actual no detecta apropiadament aquesta malaltia.
A l'Hospital Sant Joan de Déu Barcelona
Esta información es de carácter divulgativo y no sustituye la tarea de los equipos profesionales de la salud. Si necesitas ayuda, ponte en contacto con tu profesional de referencia.

Unitat de Malalties Metabòliques Congènites
Veure mésLa Unitat de Malalties Metabòliques Congènites de l'Hospital Sant Joan de Déu Barcelona col·labora amb xarxes de recerca per a oferir el millor tractament i més personalitzat possible.
Coneix-nos
